
Generative Model of Novel Inhibitor for SARS CoV 3C-like Protease
Introduction
This project is aimed to build a Graphic GAN model that would aid drug design processes. The project is also hosted on github in the following link: https://github.com/susanzhang233/mollykill. For demonstration, the project is expected to learn graphical features of the molecules that are experimentally tested with inhibition effect for the specific protein SARS coronavirus 3C-like Protease (3CLPro) . Then, the model would develop a reasonable way to generate potential novel molecules inhibitors’ graphically representations. After that, with a defeaturizer, the graphical representations would be converted into visualizable molecule formats.
Intro to GAN
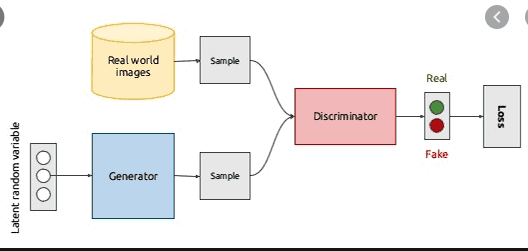
GAN, standing for Generative Adversarial Network, is commonly used in graphic works and more, such as generation of faces, music pieces. Here’s a great introduction. In this project, we’ll be building a GAN model that ultilizes the mathematical concepts of graphs in the chemical world.
Dataset
The dataset used for demonstration of this model is originally from PubChem AID1706, previously handled by JClinic AIcure team at MIT into this binarized label form. The dataset is also hosted here within this project.
Repository Explaination
model.pycontains the source codes for the GAN model createdexample.ipynbexemplifies the model usage
Demonstration:
Setup
If you are running this generator locally(i.e. in a jupyter notebook in conda, just make sure you installed:
- RDKit
- DeepChem 2.5.0 & above
- Tensorflow 2.4.0 & above
Then, please skip the following part and continue from Data Preparations.
To increase efficiency, we recommend running this molecule generator in Colab.
Then, we’ll first need to run the following lines of code, these will download conda with the deepchem environment in colab.
#!curl -Lo conda_installer.py https://raw.githubusercontent.com/deepchem/deepchem/master/scripts/colab_install.py
#import conda_installer
#conda_installer.install()
#!/root/miniconda/bin/conda info -e
#!pip install --pre deepchem
#import deepchem
#deepchem.__version__
Data Preparations
Now we are ready to import some useful functions/packages, along with our model.
Import Data
import model##our model
from rdkit import Chem
from rdkit.Chem import AllChem
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import deepchem as dc
Then, we are ready to import our dataset for training.
Here, for demonstration, we’ll be using this dataset of in-vitro assay that detects inhibition of SARS-CoV 3CL protease via fluorescence.
The dataset is originally from PubChem AID1706, previously handled by JClinic AIcure team at MIT into this binarized label form.
df = pd.read_csv('AID1706_binarized_sars.csv')
Observe the data above, it contains a ‘smiles’ column, which stands for the smiles representation of the molecules. There is also an ‘activity’ column, in which it is the label specifying whether that molecule is considered as hit for the protein.
Here, we only need those 405 molecules considered as hits, and we’ll be extracting features from them to generate new molecules that may as well be hits.
true = df[df['activity']==1]
Set Minimum Length for molecules
Since we’ll be using graphic neural network, it might be more helpful and efficient if our graph data are of the same size, thus, we’ll eliminate the molecules from the training set that are shorter(i.e. lacking enough atoms) than our desired minimum size.
num_atoms = 6 #here the minimum length of molecules is 6
input_df = true['smiles']
df_length = []
for _ in input_df:
df_length.append(Chem.MolFromSmiles(_).GetNumAtoms() )
true['length'] = df_length #create a new column containing each molecule's length
true = true[true['length']>num_atoms] #Here we leave only the ones longer than 6
input_df = true['smiles']
input_df_smiles = input_df.apply(Chem.MolFromSmiles) #convert the smiles representations into rdkit molecules
Now, we are ready to apply the featurizer function to our molecules to convert them into graphs with nodes and edges for training.
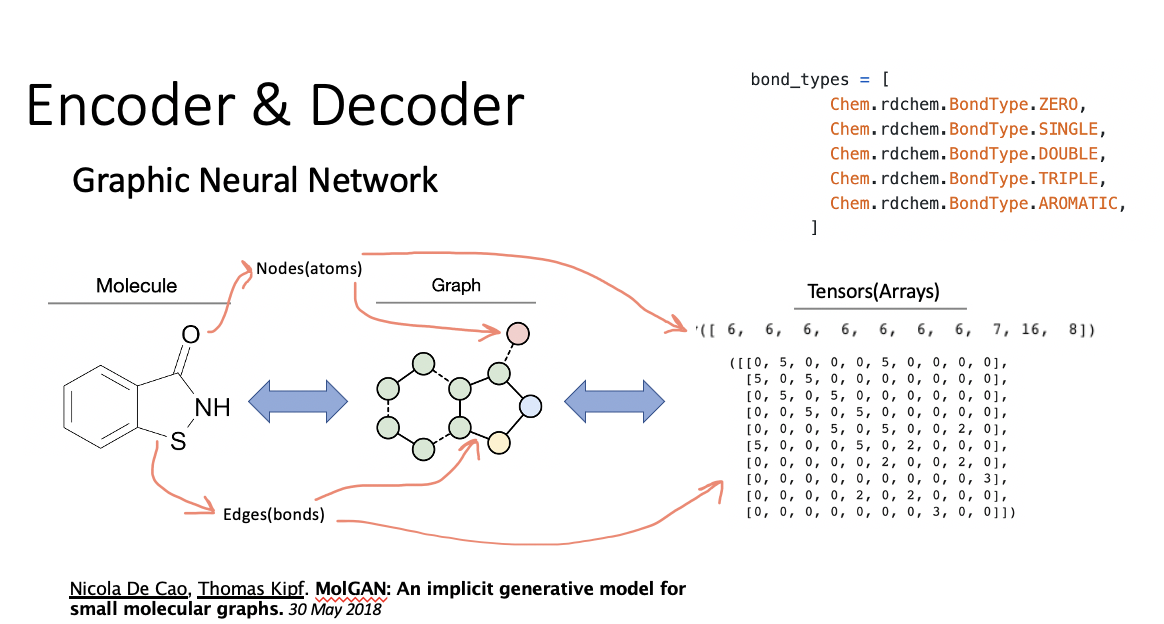 The logic behind the featurizer is to convert each molecule into an adjacency matrix storing bond informations and a node array with each atom’s features(here we only included atomic number).
The logic behind the featurizer is to convert each molecule into an adjacency matrix storing bond informations and a node array with each atom’s features(here we only included atomic number).
#input_df = input_df.apply(Chem.MolFromSmiles)
train_set = input_df_smiles.apply( lambda x: model.featurizer(x,max_length = num_atoms))
train_set
0 ([6, 6, 6, 6, 6, 8], [[0, 1, 0, 0, 0, 0], [1, ...
1 ([6, 6, 6, 6, 6, 6], [[0, 1, 0, 0, 0, 0], [1, ...
2 ([6, 6, 6, 6, 6, 6], [[0, 1, 0, 0, 0, 0], [1, ...
3 ([6, 6, 6, 6, 6, 6], [[0, 1, 0, 0, 0, 0], [1, ...
4 ([6, 6, 7, 6, 7, 6], [[0, 1, 0, 0, 0, 0], [1, ...
...
400 ([6, 6, 8, 6, 6, 8], [[0, 1, 0, 0, 0, 1], [1, ...
401 ([6, 8, 6, 8, 6, 6], [[0, 1, 0, 0, 0, 0], [1, ...
402 ([6, 8, 6, 6, 6, 6], [[0, 1, 0, 0, 0, 0], [1, ...
403 ([6, 7, 6, 6, 6, 7], [[0, 1, 0, 0, 0, 0], [1, ...
404 ([6, 6, 8, 6, 7, 16], [[0, 1, 0, 0, 1, 0], [1,...
Name: smiles, Length: 405, dtype: object
We’ll take one more step to make the train_set into separate nodes and edges, which fits the format later to supply to the model for training
nodes_train, edges_train = list(zip(*train_set) )
Training
Now, we’re finally ready for generating new molecules. We’ll first import some necessay functions from tensorflow.
import tensorflow as tf
from tensorflow import keras
from tensorflow.keras import layers
Here we’ll first initiate a discriminator and a generator model with the corresponding functions in the package.
disc = model.make_discriminator(num_atoms)
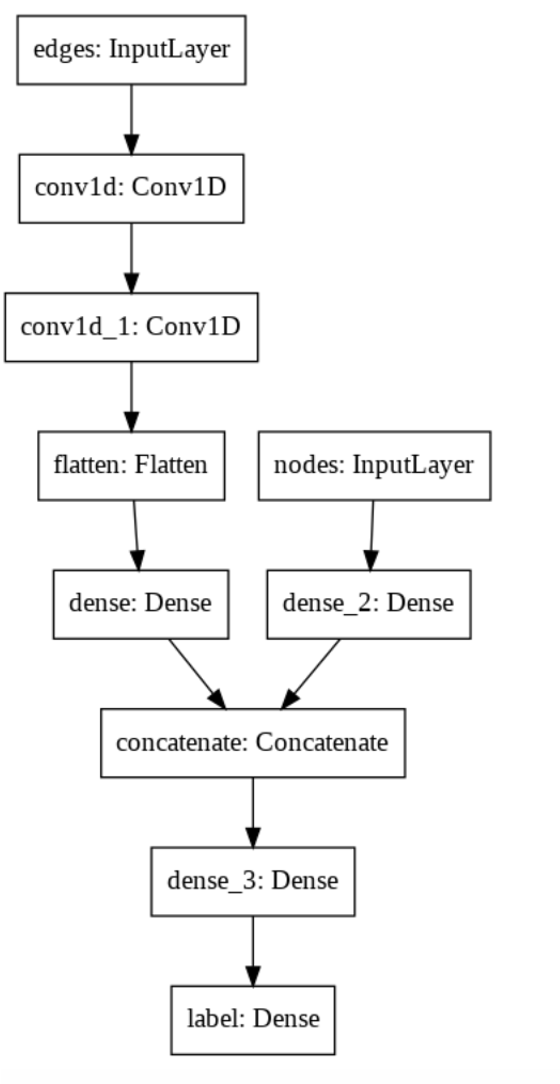
The pipeline of the discriminator is shown below:
gene = model.make_generator(num_atoms, noise_input_shape = 100)
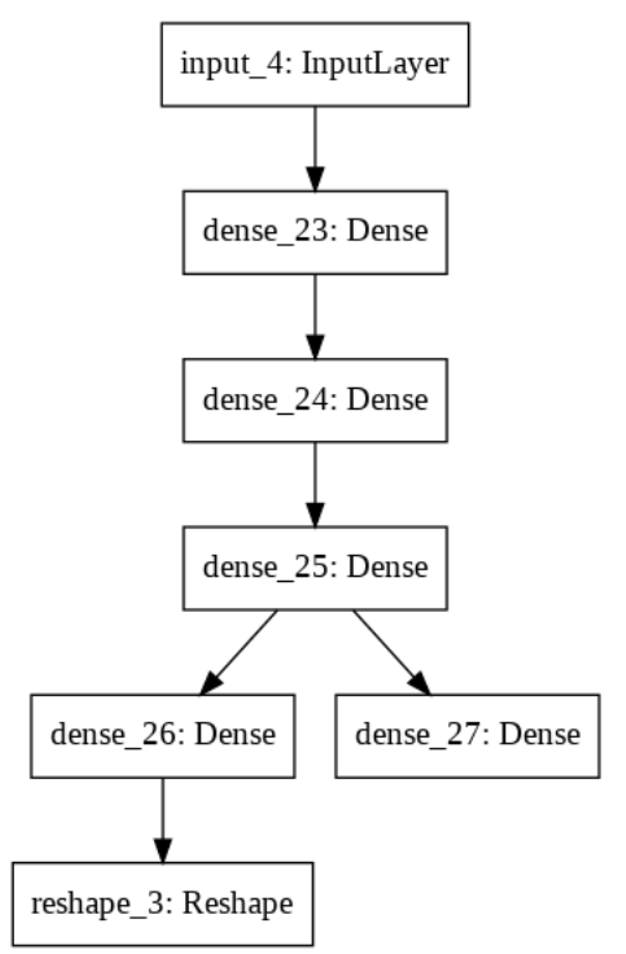
The pipeline of the generator is shown below:
Then, with the train_batch function, we’ll supply the necessary inputs and train our network. Upon some experimentations, an epoch of around 160 would be nice for this dataset.
generator_trained = model.train_batch(
disc, gene,
np.array(nodes_train), np.array(edges_train),
noise_input_shape = 100, EPOCH = 160, BATCHSIZE = 2,
plot_hist = True, temp_result = False
)
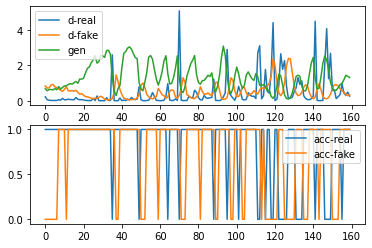
There are two possible kind of failures regarding a GAN model: model collapse and failure of convergence. Model collapse would often mean that the generative part of the model wouldn’t be able to generate diverse outcomes. Failure of convergence between the generative and the discriminative model could likely way be identified as that the loss for the discriminator has gone to zero or close to zero.
Observe the above generated plot, in the upper plot, the loss of discriminator has not gone to zero/close to zero, indicating that the model has possibily find a balance between the generator and the discriminator. In the lower plot, the accuracy is fluctuating between 1 and 0, indicating possible variability within the data generated.
Therefore, it is reasonable to conclude that within the possible range of epoch and other parameters, the model has successfully avoided the two common types of failures associated with GAN.
Rewarding Phase
The above train_batch function is set to return a trained generator. Thus, we could use that function directly and observe the possible molecules we could get from that function.
no, ed = generator_trained(np.random.randint(0,20
, size =(1,100)))#generated nodes and edges
abs(no.numpy()).astype(int).reshape(num_atoms), abs(ed.numpy()).astype(int).reshape(num_atoms,num_atoms)
(array([6, 9, 5, 7, 8, 6]),
array([[0, 0, 0, 0, 0, 0],
[1, 0, 2, 0, 0, 0],
[0, 2, 0, 2, 0, 0],
[0, 0, 2, 0, 2, 0],
[0, 0, 0, 2, 0, 3],
[2, 0, 0, 0, 3, 0]]))
With the de_featurizer, we could convert the generated matrix into a smiles molecule and plot it out=)
cat, dog = model.de_featurizer(abs(no.numpy()).astype(int).reshape(num_atoms), abs(ed.numpy()).astype(int).reshape(num_atoms,num_atoms))
Chem.MolToSmiles(cat)
'C#O=N=B=FC'
Here are some examples of molecules generated by the model. Though some molecules may appear to be not legit, a similar trend among these molecules preliminarily demonstrates that we have get the model working.
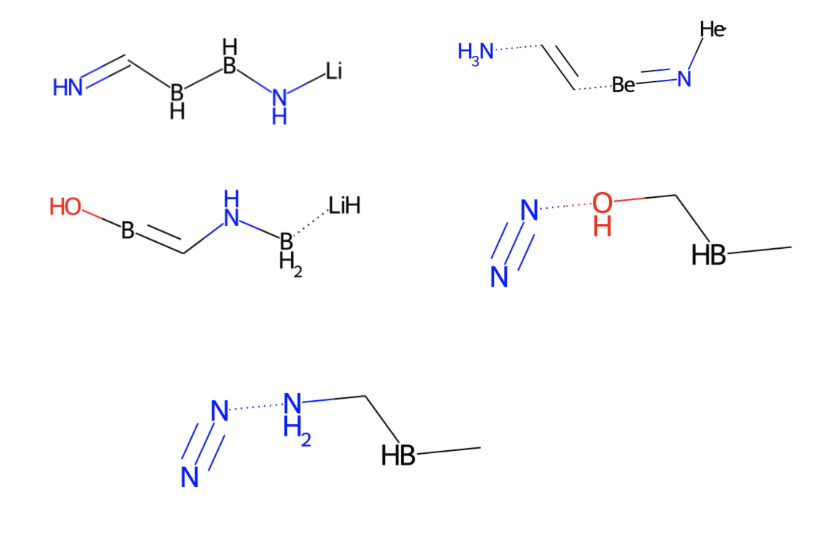
Brief Result Analysis
from rdkit import DataStructs
With the rdkit function of comparing similarities, here we’ll demonstrate a preliminary analysis of the molecule we’ve generated. With “CCO” molecule as a control, we could observe that the new molecule we’ve generated is more similar to a random selected molecule(the fourth molecule) from the initial training set.
This may indicate that our model has indeed extracted some features from our original dataset and generated a new molecule that is relevant.
DataStructs.FingerprintSimilarity(Chem.RDKFingerprint(Chem.MolFromSmiles("[Li]NBBC=N")), Chem.RDKFingerprint(Chem.MolFromSmiles("CCO")))# compare with the control
0.0
#compare with one from the original data
DataStructs.FingerprintSimilarity(Chem.RDKFingerprint(Chem.MolFromSmiles("[Li]NBBC=N")), Chem.RDKFingerprint(Chem.MolFromSmiles("CCN1C2=NC(=O)N(C(=O)C2=NC(=N1)C3=CC=CC=C3)C")))
0.017079419299743808
Limitations(Future work)
- Currently, the generation of molecules is limited to a specific length that is shorter than most molecules in the real world. Future work involving some concepts of Conditional GAN might be employed.
- The dimension of the discriminator might be improved by adding more features of the molecules(ie. hybridization, stereochemistry, etc)
- The efficiency of the featurizer in treating edges informations might be improved(viable representation of rings, etc)
Acknowledgements
- Where I’ve obtained my dataset: https://github.com/yangkevin2/coronavirus_data/tree/master/data
- A nice introduction video of GAN structure: https://github.com/whoIsTheGingerBreadMan/YoutubeVideos
- A nice introduction blogpost: https://machinelearningmastery.com/how-to-develop-a-conditional-generative-adversarial-network-from-scratch/
- MolGAN, a great previous work of related field: https://arxiv.org/pdf/1805.11973.pdf